Quantum Chemical Investigation of Spectroscopic Properties of Iron Complexes as Models for Fe-N-C Fuel Cell Catalysts
- Principal Investigators:
- Prof. Dr. Vera Krewald
- Project Manager:
- Charlotte Gallenkamp
- HPC Platform used:
- NHR4CES@TUDa: Lichtenberg Cluster Darmstadt
- Project ID:
- 01410
- Date published:
- Introduction:
- For a climate-friendly automotive sector, fuel cell technology becomes increasingly important. A promising step towards accessibility and commercialization of this technology may be reached using Fe-N-C catalyst materials for the cathode reaction of the fuel cell. With their high activity, Fe-N-C catalysts can potentially substitute currently used, expensive platinum catalysts. Fe-N-C catalysts however lack stability and their active site composition is not sufficiently well understood for systematic improvements. This project combines spectroscopy and quantum chemical calculations in order to uncover the structure of the active site(s) in Fe-N-C catalysts and better understand their electronic structures.
- Body:
-
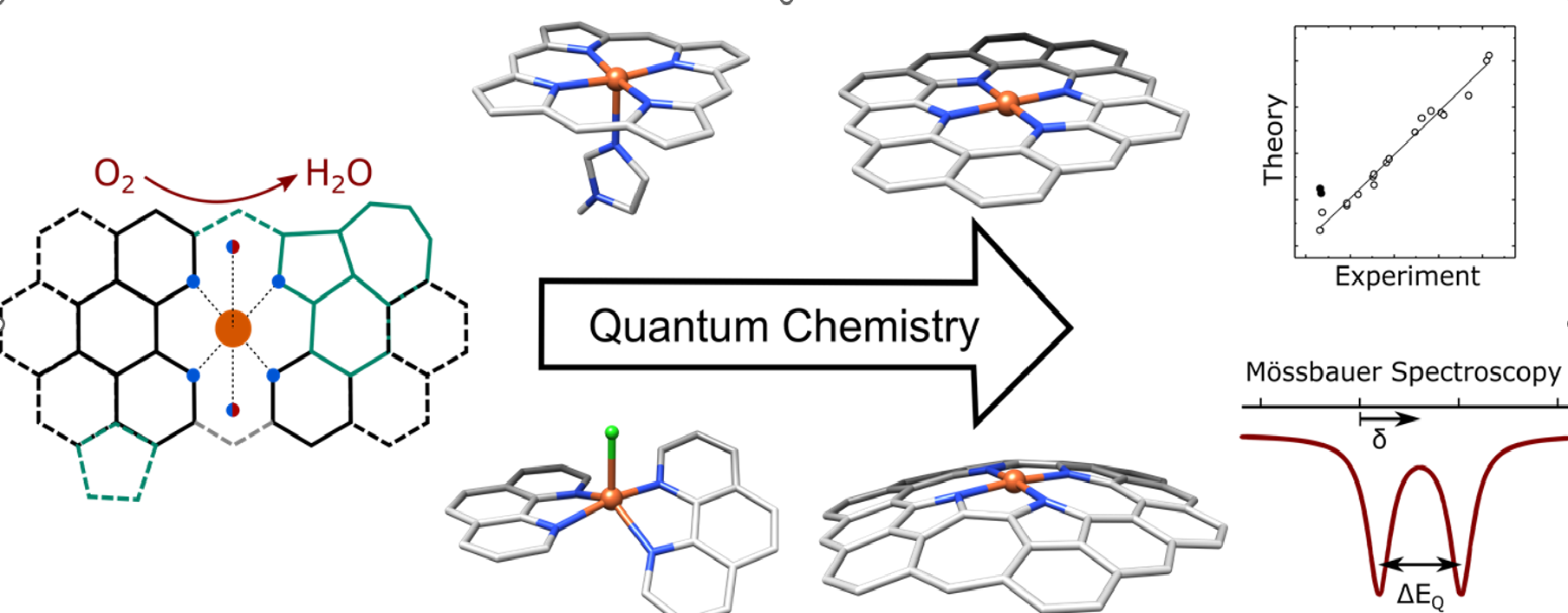
Me-N-C catalysts are a group of non-precious metal catalysts with benign, low-cost 3d transition metal centres such as manganese, iron, cobalt or copper. Catalysts of this type can activate small molecules, such as H2O, O2 and CO2 and are thus propitious for green energy research.[1] Especially for Fe-N-C catalysts, with iron as the metal centre, high activities have been reported towards oxygen reduction, a reaction taking place on the cathode side of fuel cells which has a high kinetic barrier and therefore requires large amounts of catalyst material. Using cheap and abundant Fe-N-C catalysts instead of the state-of-the-art platinum holds the promise of making fuel cell technology more accessible and thus rendering mobility more climate friendly. However, Fe-N-C catalysts do not yet fit the stability requirements, partially because there is no full understanding of the exact nature of the active site and the oxygen reduction mechanism.[2]
Fe-N-C catalysts are synthesized by pyrolysis of iron, nitrogen and carbon precursors and are assumed to have a similar active site as biologically relevant molecules of the heme family: iron is coordinated by nitrogen donor ligands, embedded in a carbon matrix. This carbon matrix is expected to comprise a more extended ?-system than the porphin backbone structure of heme molecules. Since they are pyrolyzed materials, Fe-N-C catalysts are highly amorphous and can contain functionalized carbon and iron side phases. As a consequence, not all spectroscopies are suited to study these catalysts.[2]
Mössbauer spectroscopy probes the iron nucleus and its chemical environment, and its information content is not significantly lowered by the amorphous character of a sample. By comparing the spectra of Fe-N-C catalysts to those of inorganic model complexes, it is in general possible to access the oxidation and spin state of iron and gain some information on the chemical environment of the active site.[3] However, this interpretation of Mössbauer spectra is limited to said inorganic complexes and does not consider the effect of the extended ?-system. A synthesis of such reference molecules would be difficult, but it is possible to compute characteristic parameters of Mössbauer spectra for inorganic complexes using quantum chemical methods.[4]
The project aims to design model complexes in silico that mimic the active site of Fe-N-C catalysts and to compute their Mössbauer parameters. By comparison to experiment, the active site can be identified. Using computational chemistry, it is possible to screen a large number of geometric models and to systematically study effects of small chemical changes on the spectroscopic properties of Fe-N-C catalysts. With high performance computing it is possible to calculate a large number of models in a short timeframe and to investigate models with a large number of atoms that require more computational resources.
As the computational method, density functional theory is used. During the project periods of 01023 and 01410, we performed a calibration study to estimate the accuracy of predicted Mössbauer parameters.[4] With the calculation protocol from this calibration, we investigated the influence of small chemical changes, such as dopants and axial ligands on their Mössbauer parameters, but also other spectroscopic properties were considered.[5] Molecular models were also studied as different intermediates in the oxygen reduction reaction with a special focus on their spectroscopic behaviour and a comparison to the experiment.[2] Furthermore, we systematically extended the graphene matrix to understand how this changes the electronic structure. In future, the obtained results will serve as a basis for projects in the CRC 1487 “Iron, upgraded!”.
References:
[1] A. Kumar, V. K. Vashistha, D. K. Das, S. Ibraheem, G. Yasin, R. Iqbal, T. A. Nguyen, R. K. Gupta, Md. R. Islam Fuel 2021, 304, 121420.
[2] L. Ni, C. Gallenkamp, S. Paul, M. Kübler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, V. Krewald, U. I. Kramm Adv. Energy Sustainability Res. 2021, 2, 2000064.
[3] U. I. Kramm, L. Ni, S. Wagner Adv. Mater. 2019, ?31, 1805623.
[4] C. Gallenkamp, U.I. Kramm, J. Proppe, V. Krewald Int. J. Quantum Chem. 2020, 121, e26394.
[5] C. Gallenkamp, U.I. Kramm, V. Krewald Chem. Commun. 2021, 57, 859. - Institute / Institutes:
- Fachbereich Chemie, Theoretische Chemie
- Affiliation:
- TU Darmstadt
- Image:
-