Deriving Trust Levels for Multi-Choice Data Analysis Workflows
- Principal Investigators:
- Daniel Speckhard
- Project Manager:
- Prof. Dr. Claudia Draxl
- additional Affiliation:
- SFB 1404, FONDA; NOMAD CoE; Fritz-Haber-Institut Berlin
- HPC Platform used:
- NHR@Göttingen, NHR@ZIB: HLRN Clusters Lise and Emmy
- Project ID:
- bep00098
- Date published:
- Researchers:
- Prof. Dr. Matthias Scheffler
- Introduction:
- Bringing data from various sources together, poses severe challenges to their interoperability. A prerequisite to using such data together, e.g. in machine-learning tasks, requires the assessment of the data quality. The project described here, aims at doing so by deriving trust levels for data from density-functional theory (DFT). A trust level shall be assigned for a material based on what approximation (density functional) and what numerical settings were used in the DFT simulation.
- Body:
-
Bringing data from various sources together, poses severe challenges to their interoperability. A prerequisite to using such data together, e.g. in machine-learning tasks, requires the assessment of the data quality. The project described here, aims at doing so by deriving trust levels for data from density-functional theory (DFT). A trust level shall be assigned for a material based on what approximation (density functional) and what numerical settings were used in the DFT simulation. We demonstrate our approach by computing elastic constants, band gaps, effective masses, and total energies of 1915 binary semiconductors and training statistical-learning models on this dataset.
Simulating materials properties in view of finding good candidate materials for a given application has allowed researchers and engineers to avoid wasteful trial and error in costly laboratory settings. Databases such as the Novel Materials Discovery (NOMAD, https://nomad-coe.eu) Repository invite computational scientists to upload their simulation results to an openly accessible data store. As a result, (other) researchers are able to train models on these datasets to discover new physical relationships and candidate materials [1]. These data, coming from many different sources (about 40 computer codes) are obviously extremely heterogenous, a situation that requires data-quality assessment.
This work aims at assigning trust levels to data simulated using density functional theory (DFT). A trust level shall be assigned for a material property (e.g. elastic constant, band gap, effective mass, total energy) based on what approximation (density functional) and what numerical settings were used in the DFT simulation. We choose to study binary semiconductors due to their simplicity and wide spread use in industry. DFT simulations of 1915 binary semiconductors are performed, a number that should provide sufficient training data to assess the impact of exchange-correlation functional and computational parameters on the results, and to further accurately model the effect of these settings on the convergence of these properties.
Two DFT codes, FHI-aims and exciting are employed to compute total energies, band gaps, elastic constants, and effective masses. These all-electron are representative as they employ very different basis sets. The data simulated with these precise DFT packages, will not only serve the assessment of data quality but at the same time provide a much-needed benchmark dataset. In the future, we plan to invite other DFT code developers to perform similar experiments with their code.
The research questions that this dedicated dataset will enable us to answer are: 1) What models best estimate the material property error resulting from unconverged DFT settings. 2) What can we learn from modeling the materials class of binary semiconductors in order to extend this approach to wider material classes? 3) What models allow us to reduce the uncertainty in our estimates of material property errors so as to return narrow confidence intervals.
Statistical learning models trained on these dedicated data will enable us to answer above questions and derive trust level estimates for heterogeneous data.
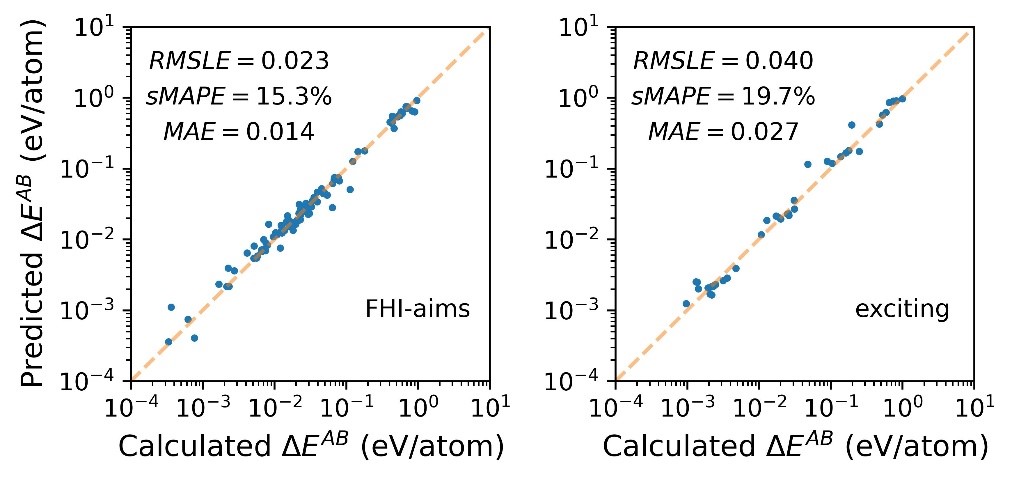
The example in Fig. 1 shows the performance of such a model [2]. In this investigation, the basis-set size was varied in the calculations of 63 binary solids with the scope of estimating the total energies in the complete basis-set limit. Based on a random-forest model, the deviation of the total energy with respect to fully converged calculations was estimated. The figure shows the relationship between the predicted and the actual error in the total energy of the binary materials (A and B being the chemical elements building up the material). This model outperforms previously used simpler linear models [3]. The data production for the 1915 materials and their properties is still ongoing.
- Institute / Institutes:
- Physics Department and IRIS Adlershof
- Affiliation:
- Humboldt-Universität zu Berlin
- Image:
-